 Home
Home
| VOLUME 95 (2012) | ISSUE 11 | PAGE 659 |
|---|
|
Consistent LDA'+DMFT - an unambiguous way to avoid double counting problem: NiO test
I. A. Nekrasov+, N. S. Pavlov+, M. V. Sadovskii+*
+Institute for Electrophysics, UB of the RAS, 620016 Ekaterinburg, Russia *Institute for Metal Physics, UB of the RAS, 620990 Ekaterinburg, Russia
Abstract
We present a consistent way of treating a double counting problem unavoidably arising within the LDA+DMFT combined approach to realistic calculations of electronic structure of strongly correlated systems. The main obstacle here is the absence of systematic (e.g. diagrammatic) way to express LDA (local density approximation) contribution to exchange correlation energy appearing in the density functional theory. It is not clear then, which part of interaction entering DMFT (dynamical mean-field theory) is already taken into account through LDA calculations. Because of that, up to now there is no accepted unique expression for the double counting correction in LDA+DMFT. To avoid this problem we propose here the consistent LDA 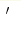 +DMFT approach, where LDA exchange correlation contribution is
explicitly excluded for correlated states (bands) during self-consistent
band structure calculations. What is left out of Coulomb interaction for those
strongly correlated states (bands) is its non-local part, which is not included
in DMFT, and the local Hartree like contribution. Then the double counting
correction is uniquely reduced to the local Hartree contribution.
Correlations for strongly correlated states are then directly accounted for via
the standard DMFT. We further test the consistent LDA+DMFT scheme
and compare it with conventional LDA+DMFT calculating the electronic structure
of NiO. Opposite to the conventional LDA+DMFT our consistent
LDA+DMFT approach unambiguously produces the insulating band
structure in agreement with experiments. +DMFT approach, where LDA exchange correlation contribution is
explicitly excluded for correlated states (bands) during self-consistent
band structure calculations. What is left out of Coulomb interaction for those
strongly correlated states (bands) is its non-local part, which is not included
in DMFT, and the local Hartree like contribution. Then the double counting
correction is uniquely reduced to the local Hartree contribution.
Correlations for strongly correlated states are then directly accounted for via
the standard DMFT. We further test the consistent LDA+DMFT scheme
and compare it with conventional LDA+DMFT calculating the electronic structure
of NiO. Opposite to the conventional LDA+DMFT our consistent
LDA+DMFT approach unambiguously produces the insulating band
structure in agreement with experiments.
|